
Malattia di Cushing
La sindrome di Cushing è una condizione rara che deriva da un eccesso di cortisolo nell’organismo. Il cortisolo è un ormone prodotto normalmente dalle ghiandole surrenali ed è fondamentale per la vita. Esso ci permette di rispondere alle situazioni di stress, ad esempio le malattie, ed ha effetti su quasi tutti i tessuti dell’organismo. Quando troppo cortisolo è prodotto dall’organismo, siamo in presenza di una sindrome di Cushing, qualunque ne sia la causa. Alcuni pazienti sviluppano la sindrome di Cushing a causa di tumore delle ghiandole surrenali che produce un eccesso di cortisolo. Altri pazienti sviluppano la sindrome di Cushing perché producono un eccesso di ACTH, ormone che stimola le ghiandole surrenali a produrre cortisolo. Solo quando l’ACTH proviene dall’ipofisi la condizione viene definita malattia di Cushing. Tumori secernenti ACTH possono originare anche in altre zone dell’organismo e vengono detti ectopici.
La sindrome di Cushing è piuttosto rara. E’ più frequente nelle donne e si manifesta tra i 20 e i 40 anni.
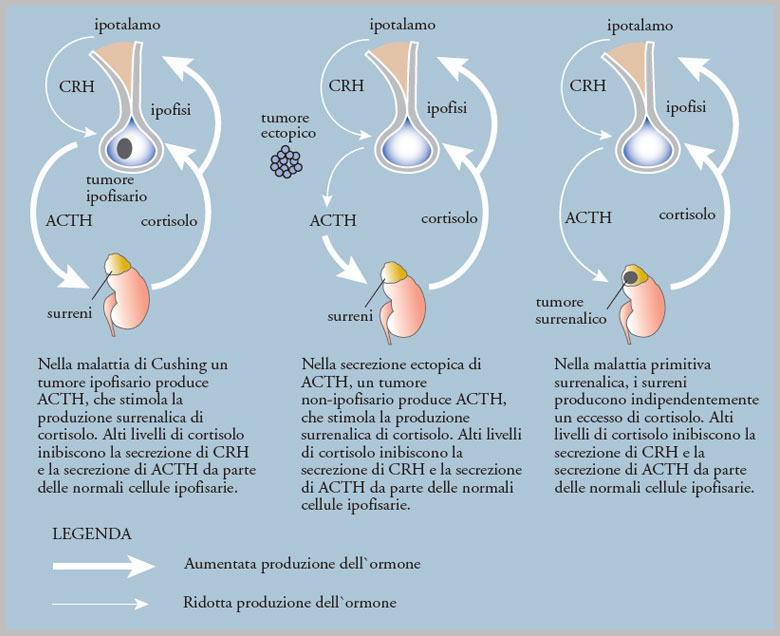
Le varie cause della sindrome di Cushing
I sintomi più comuni della sindrome sono l’aumento di peso (specialmente a livello del tronco), pressione arteriosa elevata (ipertensione), alterazioni nella memoria, nell’umore e nella concentrazione,eccesso di peluria, rossore al viso, eccesso di grasso attorno al collo, faccia rotonda,affaticamento e irregolarità mestruali.
Come viene diagnosticata la sindrome di Cushing?
Poiché non tutti i soggetti con sindrome di Cushing hanno gli stessi segni e sintomi, e poiché molti riscontri della sindrome di Cushing, come l’aumento di peso e la pressione arteriosa elevata, sono comuni nella popolazione generale, può essere difficile diagnosticare la sindrome di Cushing solo sulla base dei sintomi. Pertanto i medici usano test di laboratorio per effettuare diagnosi di sindrome e, successivamente identificare una eventuale malattia di Cushing. Questi test determinano se troppo cortisolo è prodotto spontaneamente, o se il normale sistema di controllo degli ormoni non sta funzionando correttamente. I test più comunemente usati misurano la quantità di cortisolo nella saliva o nelle urine. E’ anche possibile controllare se il cortisolo rimane elevato somministrando desametasone che riproduce l’effetto del cortisolo. Questo viene chiamato test di soppressione al desametasone. Se l’organismo sta correttamente regolando la produzione di cortisolo i livelli dimunuiscono, cosa che non accade in chi ha la sindrome di Cushing. Questi test non sempre sono in grado di diagnosticare con certezza la sindrome di Cushing perché altre patologie o problemi possono causare cortisolo in eccesso o un anomalo controllo della produzione di cortisolo. Queste condizioni che mimano la sindrome di Cushing sono chiamati stati di “Pseudo-Cushing” e comprendono disturbi psichiatrici, esercizio fisico intenso, gravidanza, stress, diabete scompensato, obesità grave, abuso di alcol, apnee del sonno A causa della somiglianza dei i sintomi e dei test di laboratorio nella sindrome di Cushing e negli stati di Pseudo-Cushing, è possibile che i medici debbano fare una serie di esami e trattare gli stati di Pseudo-Cushing – come la depressione – per vedere se i livelli elevati di cortisolo tornano nella norma durante il trattamento. Se questo non accade, è verosimile che il soggetto abbia una vera sindrome di Cushing.
Quali sono gli esami specifici per la diagnosi di sindrome di Cushing?
I pazienti con sindrome di Cushing di origine surrenalica hanno bassi livelli di ACTH nel sangue mentre pazienti con sindrome di Cushing da altra causa hanno livelli normali o elevati. Il test migliore per distinguere un tumore secernente ACTH nell’ipofisi da uno situato in un altro zona dell’organismo è una procedura chiamata cateterismo dei seni petrosi inferiori o IPSS. Esso prevede l’inserimento di cateteri nelle vene destra e sinistra della regione inguinale (o del collo) per raggiungere le vene vicino all’ipofisi. Un prelievo di sangue viene effettuato da queste sedi e da una vena periferica (braccio). Durante la procedura viene iniettato un farmaco che aumenta i livelli di ACTH. Confrontando i livelli di ACTH presenti nelle vicinanze della ghiandola ipofisi in risposta al farmaco con quelli presenti in altre parti dell’organismo è possibile fare la diagnosi.
Altri esami vengono usati per la diagnosi sindrome di Cushing: il test di soppressione al desametasone e test di simulazione con l’ormone rilasciante la corticotropina (CRH). Questi test tuttavia non sono affidabili come il cateterismo (IPSS) nel differenziare le cause. I medici di solito richiedono test multipli per confermare i risultati.
E’ anche possibile visualizzare l’ipofisi con una procedura chiamata Risonanza Magnetica (RM). Essa prevede la somministrazione di un mezzo di contrasto per facilitare la visualizzazione del tumore nell’immagine della RM. Se la RM mostra chiaramente un tumore di una certa dimensione e il test al CRH e al desametasone sono entrambi compatibili con il Cushing, può non essere necessario ricorrere al cateterismo. Esiste però una percentuale (fino al 10%) della popolazione sana che mostra un’area anomala nell’ipofisi compatibile con un tumore. Pertanto, la presenza di una anomalia singola non è diagnostica di sindrome di Cushing. Inoltre, nel 50% circa dei pazienti con malattia di Cushing, il tumore è troppo piccolo per essere rilevato. Così l’assenza di un tumore alla RM non esclude necessariamente la malattia di Cushing.
Qual è la terapia per la sindrome di Cushing?
Gli unici trattamenti efficaci per la sindrome di Cushing sono l’asportazione del tumore, la riduzione dalla sua capacità di produrre ACTH, o l’asportazione delle ghiandole surrenali. Ci sono altri approcci complementari che possono essere usati per trattare alcuni dei sintomi. Per esempio il diabete, la depressione e l’ipertensione arteriosa sono trattati con le terapie abituali. I medici possono prescrivere anche un integrazione di calcio e vitamina D, o altre terapie per prevenire l’assottigliamento dell’ossa. La rimozione del tumore ipofisario è il modo migliore per trattare la malattia di Cushing. Ciò è raccomandato in coloro che hanno un tumore che non si estende al di fuori dell’area ipofisaria e che sono in buone condizioni per sottoporsi all’ anestesia. L’intervento è effettuato di solito passando per naso o il labbro superiore e attraverso il seno sfenoidale per raggiungere il tumore. Questa è la cosiddetta chirurgia trans-sfenoidale ed evita di dover raggiungere l’ipofisi passando per la volta del cranio. La rimozione del tumore lascia il resto della ghiandola ipofisaria intatto così che alla fine funzionerà normalmente. La percentuale di successo è del 70- 90% quando l’intervento è eseguito da un chirurgo esperto. La percentuale di successi riflette l’esperienza del chirurgo che esegue l’intervento. Il tumore può tuttavia recidivare nel 15% dei pazienti, probabilmente a causa dell’incompleta rimozione del tumore all’interevento iniziale. Le terapie mediche comunemente usate per curare la sindrome di Cushing sono il ketoconazolo che è un farmaco antifungino che inibisce la produzione di ormoni surrenalici (e quindi anche di cortisolo) oppure il mitotano che è un farmaco chemioterapico che provoca la distruzione delle cellule surrenaliche ed è pertanto maggiormente utilizzato nei carcinomi surrenalici (con o senza sindrome di Cushing associata). Per ciò che riguarda la malattia di Cushing l’unica terapia approvata al momento attuale è il pasireotide, farmaco che agisce sulle cellule ipofisarie che possiedono recettori per la somatostatina bloccando sia la crescita tumorale che la produzione ormonale.
L’asportazione di entrambe le ghiandole surrenali elimina la capacità dell’organismo di produrre cortisolo e rappresenta un’ opzione terapeutica successiva alla chirurgia tran sfenoidale e alla terapia medica. Poiché gli ormoni surrenali sono indispensabili per la vita, i pazienti dovranno assumere un farmaco simile al cortisolo ed all’ ormone aldosterone, che controlla l’equilibrio dei sali minerali e dell’acqua, tutti i giorni per il resto della loro vita.
Altre opzioni di trattamento includono l’irradiazione dell’intera ghiandola o la radioterapia diretta sul bersaglio (chiamata radiochirurgia), quando il tumore è visibile in RM. Questo può essere usato come trattamento unico oppure nel caso in cui la chirurgia ipofisaria non abbia avuto completo successo. Queste metodiche possono richiedere fino a 10 anni per avere un effetto completo.
Nel frattempo i pazienti devono assumere farmaci per ridurre la produzione surrenalica di cortisolo. Un importante effetto collaterale della radioterapia è che può colpire le altre cellule ipofisarie che producono ormoni differenti. Come conseguenza, fino al 50% dei pazienti ha la necessità di assumere una terapia sostitutiva ormonale entro dieci anni dal trattamento.
Un endocrinologo esperto di ipofisi e di neuro-endocrinologia può decidere la migliore strategia di trattamento.