
Tumori Neuroendocrini (NET)
I tumori neuroendocrini (NET) sono comunemente ritenuti rari, anche se la loro incidenza negli ultimi anni è aumentata (35 casi/100000/anno); nel 70% dei casi sono ben differenziati e a basso grado di malignità, e caratterizzati da lunghe sopravvivenze. Nel caso producano sostanze ormonali, questi tumori possono presentarsi con sintomi invalidanti come la diarrea cronica refrattaria, flushing (rossore del volto) e cefalea. Nel 20% circa dei casi si tratta invece di tumori francamente maligni, che metastatizzano prevalentemente al fegato. I rimanenti tumori sono indifferenziati, di solito biologicamente asintomatici, con prognosi sempre infausta a breve termine.
I NET sono una famiglia eterogenea di tumori che comprende:
1. Tumori neuroendocrini gastro-entero-pancreatici (GEP-NET):
• Insulinoma
• Gastrinoma:
– Tipo I associato alla gastrite trofica autoimmune ed ipergastrinemia
– Tipo II sindrome di Zollinger-Ellison in MEN1
– Tipo III o sporadico, caratterizzato da una maggiore aggressività
• Somatostatinoma
• Glucagonoma
• VIPoma
• NET secernenti PP (polipeptide pancreatico)
• NET non funzionanti
2. Carcinoidi bronchiali
3. Carcinoidi timici
4. Tumore a Cellule di Merkel
5. Feocromocitoma/Paraganglioma
6. Adenoma ipofisario, surrenalico e paratiroideo
7. Carcinoma midollare della tiroide
I NET possono essere sporadici o con carattere di malattia familiare, diagnosticata in pazienti che appartengono a famiglie note per disordini ormonali (MEN-Neoplasie Endocrine Multiple).
Lo spazio occupato da questi tumori è andato sempre più aumentando negli ultimi anni, trasformandosi in un problema col quale il clinico deve iniziare a misurarsi anche (e soprattutto) al di fuori dei grossi centri di riferimento oncologico. Questo fatto ha portato molti pazienti a rivolgersi anche a piccoli centri, il più delle volte carenti di dotazioni tecnologiche e culturali adeguate. L’aumento del numero di casi diagnosticati negli ultimi anni rende ragione dell’attuale maggiore attenzione da parte del clinico nei riguardi di questa particolare patologia, con impiego di alcune nuove tecniche diagnostiche di radiodiagnostica e di laboratorio, e con l’uso terapeutico di nuove molecole, anche con approccio radiometabolico, con un impatto favorevole sul decorso della malattia.
E’ di fondamentale importanza però, che questi pazienti siano gestiti da centri di eccellenza per i tumori neuroendocrini (www.netroma.org), costituiti da team multidisciplinari, così da assicurare al paziente il percorso diagnostico-terapeutico più appropriato.
 Team multidisciplinare del Policlinico Gemelli
Team multidisciplinare del Policlinico Gemelli
Centro di eccellenza ENETS Roma
Policlinico Gemelli – Azienda Ospedaliera Sant’Andrea
Il Centro di Eccellenza ENETS di Roma dispone, presso ciascuna delle due sedi (Azienda Ospedaliera Sant’Andrea e Policlinico Gemelli), di un team mutlidisciplinare di esperti, che collaborano nella gestione dei pazienti affetti da Tumore Neuroendocrino. I due team si riuniscono mensilmente in un unico board multidisciplinare per la discussione e la condivisione dei casi clinici. Il centro ENETS di Roma è attivo dal 2014 presso l’Azienda Ospedaliera Sant’Andrea ed il Policlinico Gemelli, associati in una stessa unità funzionale, rappresentando un riferimento per i pazienti affetti da Tumore Neuroendocrino. I responsabili del Centro sono, per il Policlinico Gemelli, il Prof. Guido Rindi, e per l’Azienda Ospedaliera Sant’Andrea, il Prof. Gianfranco Delle Fave.
 Certificato Centro di Eccellenza ENETS – Consegna del certificato del Centro di Eccellenza durante il Congresso ENETS 2014 (Barcellona 5-7 Marzo 2014)
Certificato Centro di Eccellenza ENETS – Consegna del certificato del Centro di Eccellenza durante il Congresso ENETS 2014 (Barcellona 5-7 Marzo 2014)
Diagnosi
I NET vengono divisi in due distinte categorie, funzionanti e non funzionanti, in relazione alla capacità di produrre e secernere sostanze ormonali. La diagnosi di laboratorio è utile nel tumore funzionante. Essa prevede la ricerca nel plasma di ormoni prodotti dalla neoplasia e/o di altri marcatori specifici di malattia. La ricerca di markers neoplastici come gastrina ed insulina plasmatiche ed acido 5-idrossindolacetico urinario (5-HIAA), per esempio, aiuta il clinico ad orientarsi nella diagnosi. In particolare, la gastrina si presenta elevata nel plasma nel gastrinoma, l’insulina nell’insulinoma ed il 5-HIAA, catabolita urinario della serotonina, si può osservare a livelli molto elevati nelle urine dei pazienti affetti da sindrome da carcinoide. Altri marcatori, sono l’ enolasi neurono-specifica (NSE) e la cromogranina A. Per quanto riguarda la diagnosi strumentale, di fondamentale importanza, oltre alla TAC ed alla RMN, c’è la PET (positron emission tomography) con 68-Ga, con 18F-DOPA, con 11C-5-HTTP e con 18-FDG, e l’ecoendoscopia (EUS).
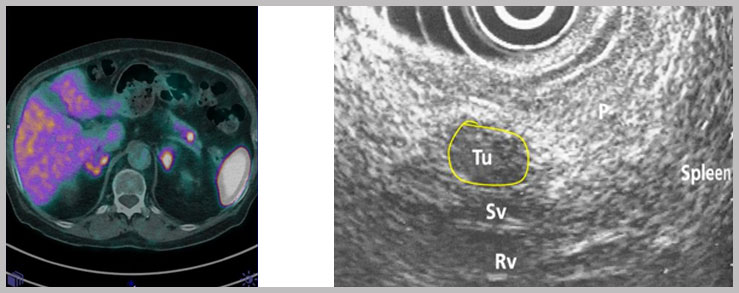
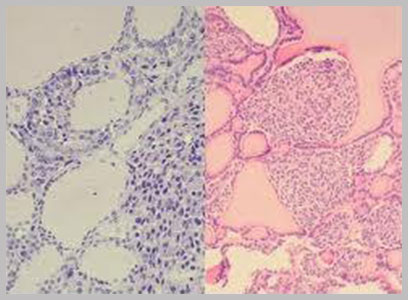
È necessario precisare che le tecniche radiodiagnostiche, sia morfologico sia funzionale, non dà informazioni sul grado di differenziazione o sulla secrezione ormonale specifica del tumore, che invece ci dà la diagnosi immuno-istopatologica, in grado di dare informazioni anche in termini prognostici, dosando l’espressione di alcune proteine (NSE, cromogranina A, sinaptofisina, cheratine), alcune delle quali legate all’angioinvasività e alla proliferazione cellulare (ki67), permettendo così di studiare la malattia allo scopo di garantire un più corretto approccio terapeutico.
L’identificazione del fenotipo neuroendocrino prevede l’impiego di specifici marcatori neuroendocrini. Tra questi, Cromogranina A (CgA) e Sinaptofisina sono i più attendibili per sensibilità e specificità. Le forme tumorali poco differenziate possono mostrare espressione di marcatori neuroendocrini ridotta o assente. Di fondamentale importanza è anche l’indice mitotico, che insieme al Ki67 consente di classificare il tumore in tre principali categorie:
• NET G1: tumore neuroendocrino G1 (< 2 mitosi o Ki67 <2%)
• NET G2: tumore neuroendocrino G2 (2-20 mitosi o Ki67 3-20%)
• NEC G3: carcinoma neuroendocrino G3 (> 20 mitosi o Ki67 > 20%)
Terapia
Negli ultimi anni è sempre più diffuso l’uso, come trattamento di prima linea oltre della chirurgia, degli analoghi della somatostatina (octreotide, lanreotide). Altri approcci terapeutici sono la terapia radiorecettoriale (PRRT), le target therapy (everolimus e sunitinib) e nei casi di carcinoma neuroendocrina (NEC), la chemioterapia.